


Microsoft's BioEmu: Transforming Protein Dynamics with AI-Driven Equilibrium Simulations
Microsoft’s BioEmu model is redefining protein dynamics with unprecedented efficiency, and it’s open-sourced now.

Introduction
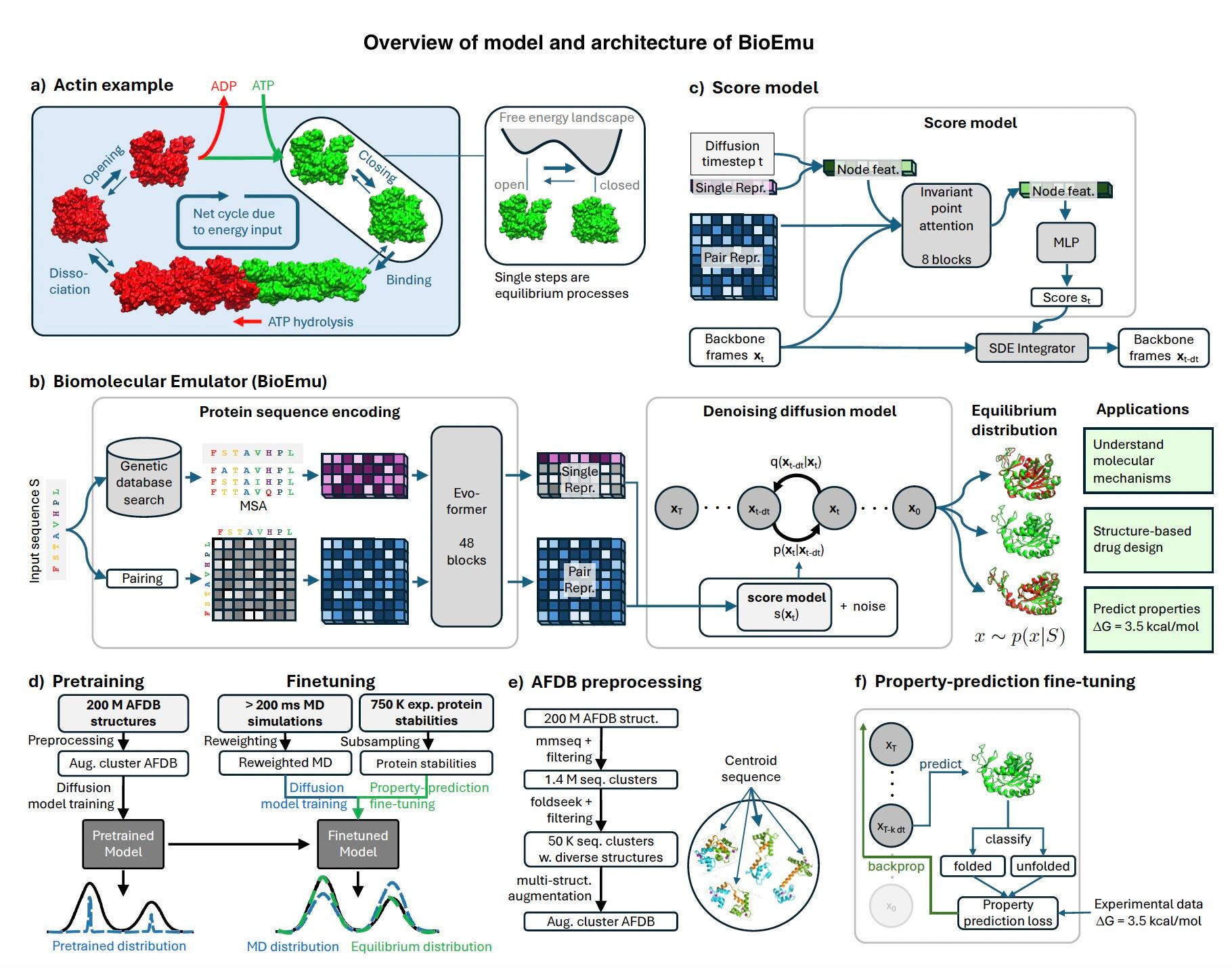
Understanding protein dynamics is fundamental to drug discovery, enzyme engineering, and biomaterials research. However, traditional molecular dynamics (MD) simulations, while effective, are computationally intensive and often impractical for large-scale studies. Microsoft’s BioEmu, a generative deep learning model, offers a scalable solution by efficiently recreating protein equilibrium ensembles at a fraction of the computational cost.
BioEmu leverages advanced AI methodologies to generate thousands of equilibrium protein structures per hour on a single GPU, making it a powerful alternative to conventional simulation techniques. The model’s ability to capture biologically relevant protein motions, cryptic pocket formations, and local unfolding transitions positions it as a transformative tool in molecular simulation.
Overcoming Computational Barriers in Protein Simulations
Efficiency and Speed
Traditional MD simulations require weeks of high-performance computing to capture equilibrium conformations.
BioEmu achieves comparable results within hours on a single GPU, significantly reducing computational demands.
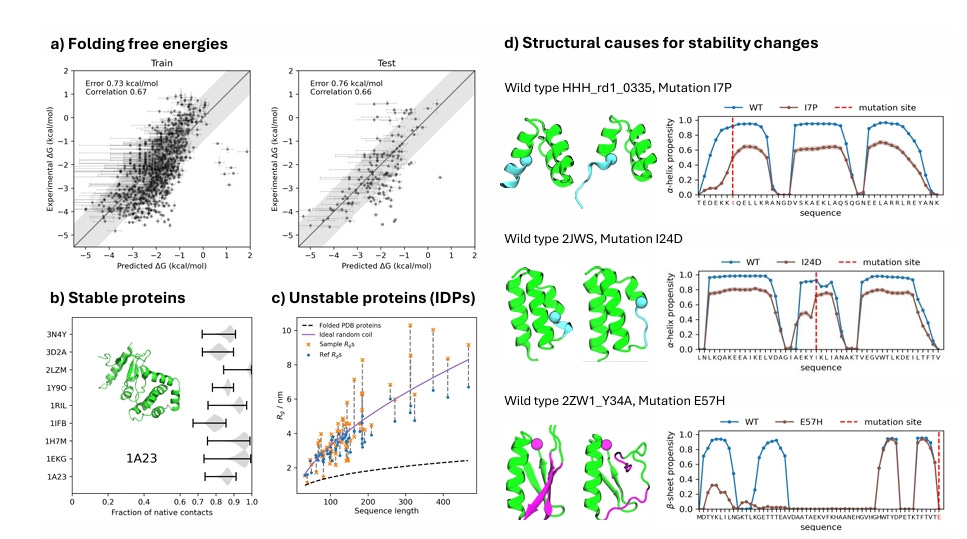
Accuracy in Free Energy Landscapes
The model predicts protein stability and conformational states with errors below 1 kcal/mol, rivaling high-precision MD simulations.
By incorporating extensive molecular simulation datasets and experimental stability measurements, BioEmu ensures high fidelity in thermodynamic predictions.
Capturing Functionally Relevant Protein Motions
BioEmu can identify cryptic binding pockets, an essential factor in drug discovery.
The model enables realistic sampling of protein conformational states, offering insights into structural flexibility and ligand-binding mechanisms.
Cost-Effective Alternative to Conventional Approaches
BioEmu eliminates the need for long-timescale simulations, making large-scale protein studies feasible without requiring extensive computational resources.
Core Methodology: AI-Powered Protein Conformational Sampling
BioEmu utilizes a generative deep learning framework trained on a combination of:
Over 200 milliseconds of MD simulations, covering a wide range of protein folding events and stability variations.
Experimental protein stability data, ensuring thermodynamic accuracy in sampled conformations.
Machine learning-enhanced structure prediction, integrating advances from AlphaFold and diffusion models to improve sampling diversity and accuracy.
A key innovation in BioEmu’s architecture is Property-Prediction Fine-Tuning (PPFT), which enhances the model’s ability to predict experimental observables such as protein stability. By integrating partial backpropagation techniques, BioEmu optimizes training efficiency while maintaining high predictive accuracy.
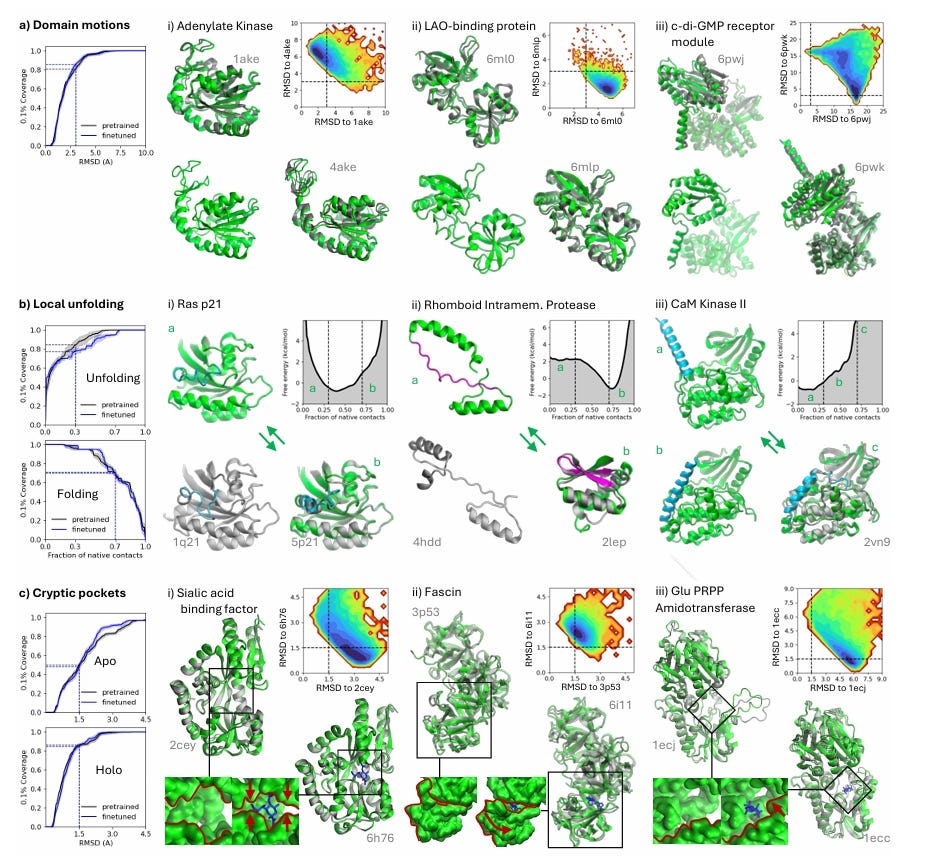
Applications in Drug Discovery and Structural Biology
Advancing Drug Discovery
BioEmu enables researchers to predict ligand-induced conformational changes, identifying potential allosteric sites and cryptic pockets that may not be evident in static protein structures.
The model facilitates high-throughput virtual screening, improving hit identification in computational drug design.
Enhancing Protein Engineering
The ability to accurately predict protein stability and domain flexibility supports rational protein engineering for enzyme design and therapeutic development.
BioEmu serves as a computationally efficient alternative to traditional mutagenesis experiments, accelerating protein optimization workflows.
Integrating AI with Experimental Structural Biology
By generating equilibrium ensembles comparable to those obtained through cryo-electron microscopy (cryo-EM) and X-ray crystallography, BioEmu aids in experimental data interpretation and structural validation.
The model’s ability to predict free energy landscapes with high accuracy provides insights into protein folding mechanisms and stability variations across different experimental conditions.
Conclusion
BioEmu represents a significant advancement in AI-driven molecular simulations, addressing the scalability limitations of traditional molecular dynamics simulations. By enabling the rapid and accurate generation of protein equilibrium ensembles, BioEmu paves the way for more cost-effective, high-throughput, and precise molecular studies.
The introduction of machine learning-based equilibrium simulations marks a critical step toward integrating AI-driven approaches with experimental and computational biophysics. As machine learning continues to evolve, models like BioEmu will play an increasingly pivotal role in rational drug design, protein engineering, and biomolecular research.
This advancement redefines the future of protein simulations, making high-precision conformational sampling and thermodynamic predictions accessible at an unprecedented scale.
🔗 Check out the open-source repository here: https://github.com/microsoft/bioemu
📜 Check out the paper: https://www.biorxiv.org/content/10.1101/2024.12.05.626885v1.abstract